 全国免费服务热线:020-83487199
全国免费服务热线:020-83487199
 全国免费服务热线:020-83487199
全国免费服务热线:020-83487199

写在前面
在过去的30年里,药物的稳定性实验已从仅仅检测产品中有害物质的变化发展到运用科学原理对药品进行综合性的质量控制。目前,世界各国对此项工作逐渐重视,各国都已开始制定适合本国的药品稳定性实验规范。由于稳定性研究周期较长,因此从建立方案时就应对产品详尽分析,还应考虑国际协调指南(ICH指南)、欧洲药物审评机构要求和美国FDA稳定性实验要求等。按照这些指南或要求所获得的稳定性数据将有利于药品的全球化注册。


今天这篇文章,将为大家带来有关“稳定性试验”的一些名词解释,全篇干货,建议直接收藏噢~
药物的稳定性:在特定的容器或密闭系统中可保持药物原有的物理、化学、生物学、治疗、毒理及其相关性能。最理想的情况是药物原料的纯度、杂质含量在各个安全性实验、临床试验评估、制剂研究和稳定性实验中都保持恒定。
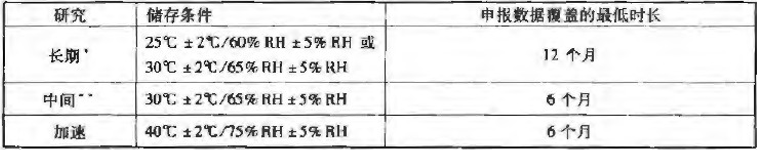
加速试验(Accelerated testing):加速试验是采用超出贮藏条件的试验设计来加速原料药或制剂的化学降解或物理变化的试验,是正式稳定性研究的一部分。(加速试验数据还可用于评估在非加速条件下更长时间的化学变化,以及在短期偏离标签上注明的贮藏条件(如运输过程中)时对质量产生的影响;但是,加速试验结果有时不能预测物理变化。)
中间试验或中间条件试验:中间试验是为拟在25℃下长期贮藏的原料药或制剂设计的在30℃/65%RH条件下进行的试验,目的是适当加速原料药或制剂的化学降解或物理变化。
长期试验:长期试验是为确定在标签上建议(或批准)的有效期(复检期)进行的,在拟定贮藏条件下的稳定性研究。
注册批次:用于正式稳定性研究的原料药或制剂批次,其稳定性数据在注册申报时可分别用于建立原料药和制剂的有效期(复检期)。原料药申报批次均至少是中试规模;新制剂3个批次中至少2个批次是中试规模,另1个批次的规模可小一些,但必须采用有代表性的关键生产步骤;仿制制剂申报批次均至少是中试规模。注册批次也可以是生产批次。
生产批次:使用申报时确认的生产厂房及生产设备,以生产规模生产的原料药或制剂批次。
承诺批次:注册申报时承诺的在获得批准后开始进行或继续完成稳定性研究的原料药或制剂的生产规模批次。


复检期:通常对多数已知不稳定的生物技术/生物原料药和某些抗生素,建立确认的是有效期,而对多数较稳定的化学原料药,建立确认的实为复检期。复检期是在此期间内,只要原料药保存于规定的条件下,就认为其符合质量标准,并可用于生产相应的制剂;而在此期限后,如果用该批原料药生产制剂,则必须进行质量符合性复检;如复检结果显示其质量仍符合质量标准,则应立即使用;1批原料药可以进行多次复检,且每次复检后可以使用其中的一部分,只要其质量一直符合质量标准即可。
影响因素试验(制剂):是指为评估剧烈条件对制剂质量的影响而进行的研究。该试验包括光稳定性试验和对某些制剂(如:定量吸入制剂、乳膏剂、乳剂和需冷藏的水性液体制剂)的特定试验。



 当前位置:
当前位置:

 招贤纳士
招贤纳士 资料下载
资料下载 服务网点
服务网点 联系方式
联系方式












 复制网址
复制网址 邮件
邮件 QQ空间
QQ空间 新浪微博
新浪微博 腾讯微博
腾讯微博 微信
微信 人人网
人人网 搜狐微博
搜狐微博 QQ好友
QQ好友 开心网
开心网 飞信
飞信 豆瓣
豆瓣 一键分享
一键分享 更多
更多